Digital Discovery, 2023, 2,1233-1250
DOI: 10.1039/D3DD00113J, Perspective
DOI: 10.1039/D3DD00113J, Perspective
 Open Access
Open Access This article is licensed under a Creative Commons Attribution 3.0 Unported Licence.
This article is licensed under a Creative Commons Attribution 3.0 Unported Licence.Kevin Maik Jablonka, Qianxiang Ai, Alexander Al-Feghali, Shruti Badhwar, Joshua D. Bocarsly, Andres M. Bran, Stefan Bringuier, L. Catherine Brinson, Kamal Choudhary, Defne Circi, Sam Cox, Wibe A. de Jong, Matthew L. Evans, Nicolas Gastellu, Jerome Genzling, María Victoria Gil, Ankur K. Gupta, Zhi Hong, Alishba Imran, Sabine Kruschwitz, Anne Labarre, Jakub Lála, Tao Liu, Steven Ma, Sauradeep Majumdar, Garrett W. Merz, Nicolas Moitessier, Elias Moubarak, Beatriz Mouriño, Brenden Pelkie, Michael Pieler, Mayk Caldas Ramos, Bojana Ranković, Samuel G. Rodriques, Jacob N. Sanders, Philippe Schwaller, Marcus Schwarting, Jiale Shi, Berend Smit, Ben E. Smith, Joren Van Herck, Christoph Völker, Logan Ward, Sean Warren, Benjamin Weiser, Sylvester Zhang, Xiaoqi Zhang, Ghezal Ahmad Zia, Aristana Scourtas, K. J. Schmidt, Ian Foster, Andrew D. White, Ben Blaiszik
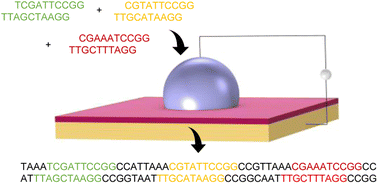
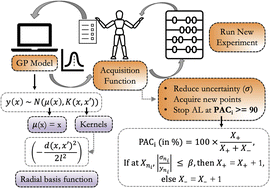
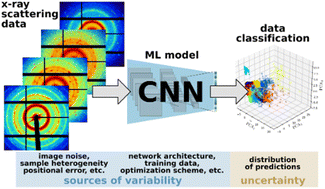
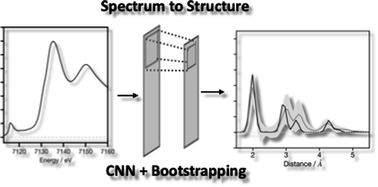
We report the findings of a hackathon focused on exploring the diverse applications of large language models in molecular and materials science.
The content of this RSS Feed (c) The Royal Society of Chemistry
We report the findings of a hackathon focused on exploring the diverse applications of large language models in molecular and materials science.
The content of this RSS Feed (c) The Royal Society of Chemistry